About
Mission
News
Events
Contact Us
People
Faculty
Postdoctoral Scholars
Graduate Students
Administrative Staff
Research Staff
Research
Publications
Computational Neuroscience Center
Keck Imaging Center
Resources
Global Community
Trainees
Equity, Diversity & Inclusion
NBIO Intranet
International Travel Registry
Give
LinkedIn
Bluesky
Instagram
Faculty
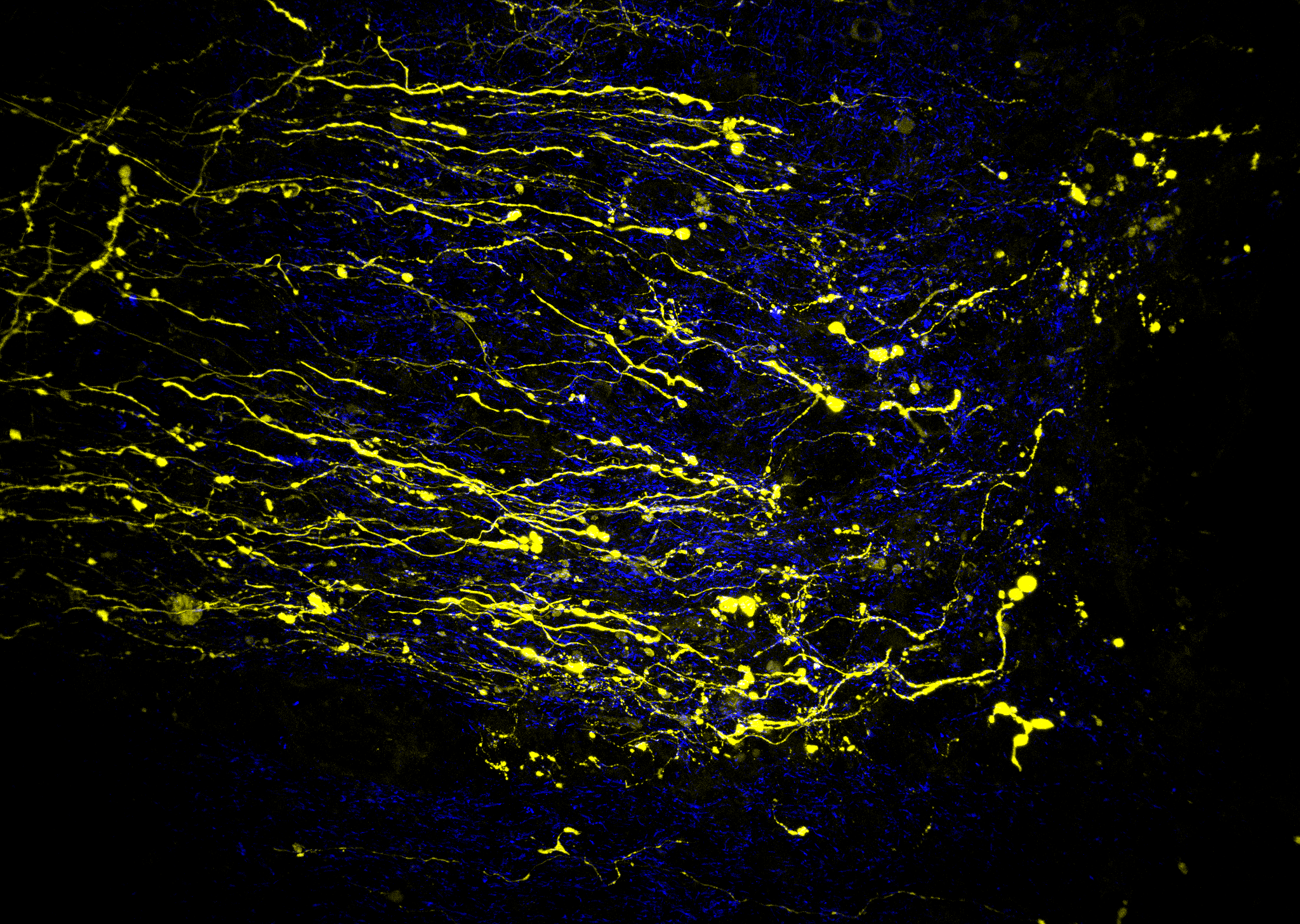
Image:
Dystrophic Corticospinal Rat Neurons Following Spinal Cord Injury – Blue- Nathaniel Peters